Assembling the pieces
Contents
8. Assembling the pieces#
In this chapter, no new Python topics will be introduced. You will practice by writing functions that combine the different topics that we have covered.
8.1. Exercises#
Exercise 8.1: Translation
Write a function mrna2protein that converts an mRNA sequence (using the A,C,G,T alphabet) into
a protein sequence.
You can use the codon table that was used in exercise 5.9.
def mrna2protein(seq):
# Adapt this function with your own code
return prot
Exercise 8.2: Open reading frames
Write a function longest_orfs that will take a DNA sequence as input and returns the protein sequence encoded by
the longest ORF for all three frames. The return value should be a dictionary, with the frame number as the key and the
protein sequence as the value. This means that the dictionary will have three key:value pairs. The three keys are 1,
2 and 3 and the corresponding values are the longest ORF for frame 1, the longest ORF for frame 2 and the longest ORF for frame 3.
The stop codon (* in the amino acid table) should not be included in the translation of the ORF. For this exercise an open reading frame does not need to start with an ATG, it can start with any codon, except a stop codon.
For example, take the sequence "actgcgtagagagctggagagattaggc". The translation of the full sequence for frame 1 would look like this:
TA*RAGEIR
In this example, the longest ORF is RAGEIR.
You can re-use code that you have written for exercise 4.8 and exercise 8.1.
def longest_orfs(seq):
# Adapt this function with your own code
return orfs
Exercise 8.3: Hamming distance
Write a function called hamming that takes two strings s and t of equal length as arguments and computes the number of differences between them.
The function should return the number of symbols that differ in s and t.
def hamming(s, t):
# adapt this function with your own code
print(hamming("ACTG", "ACTG"))
print(hamming("ACTG", "GTCA"))
print(hamming("AACC", "AATT") + hamming("CTGA", "TCGA"))
If your function is defined correctly, the code above should exactly match the following output when run:
0
4
4
Exercise 8.4 Find exact motif matches
Given two strings s and t, t is a substring of s if t
is contained as a contiguous collection of symbols in s (as a result,
t must be no longer than s).
Write a function called find_match that takes two arguments, s and t,
and returns a list of all positions of the substring t in s.
# define your function here
When you have finished the function run the code below:
# Don't change anything here, but update the function definition above!
print(find_match("GATATATGCATATACTT", "ATAT"))
print(find_match("AUGCUUCAGAAAGGUCUUACG", "U"))
Instead of giving an error, the output above should exactly match the following:
[2, 4, 10]
[2, 5, 6, 15, 17, 18]
Exercise 8.5: Motif conversion
Write a function that converts a consensus sequence into a positional weight matrix.
The consensus sequence can be composed of symbols from the IUPAC DNA code.
The function should ignore any non-IUPAC character. The positional weight matrix should be a two-dimensional list.
Every element in the first list is a list containing the relative frequencies of A, C, G and T, in that specific order, that together sum up to 1.0.
The function should work, regardless of the input being upper-case, lower-case or a mix.
You may copy-paste the following dictionary as a starting point of your function:
iupac_dict = {
'A':['A'],
'C':['C'],
'G':['G'],
'T':['T'],
'U':['U'],
'R':['A', 'G'],
'Y':['C', 'T'],
'S':['G', 'C'],
'W':['A', 'T'],
'K':['G', 'T'],
'M':['A', 'C'],
'B':['C', 'G', 'T'],
'D':['A', 'G', 'T'],
'H':['A', 'C', 'T'],
'V':['A', 'C', 'G'],
'N':['A', 'C', 'G', 'T']}
You may not use a dictionary with predefine values, e.g. {'V': [0.33, 0.33, 0.33, 0], etc}, in your answer.
If you are struggling with this exercise, click on the triangle to view tips.
To do this exercise, you need to know what the IUPAC DNA code is, and what a positional weight matrix (PWM) is. These concepts are covered in lecture. Review the lecture materials before you move forward with this exedrcise.
Let’s take the following IUPAC consensus sequence:
TGASTCAAs you know, DNA contains only 4 nucleotides: A, C, G, and T. In the consensus sequence above, there is a letter “S”. What does “S” mean?
In the lecture, we covered 3 different ways to write a PWM in Python. Two of these are two-dimensional lists (also known as “list of lists”), and one is a dictionary. Go to the lecture slide with the three versions of PWMs. Which two PWMs are two-dimensional lists, and which one is a dictionary?
Take the following PWM:
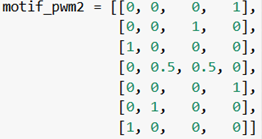
As you can see, the ordering of this PWM is not explicitly defined. What is the implicit/default ordering of PWMs written in this format?
The IUPAC consensus sequence given in part 1, and the PWM given in part 3, contain the same information. Can you explain how?
If you’re given the IUPAC consensus sequence in part 1, what steps would you take to end up with the PWM in part 3?
Take the IUPAC consensus sequence one letter at a time. For the first letter (T), you should end up with:
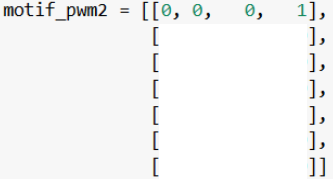
(Optional!) Below is another way to write a PWM as a two-dimensional list. If you’re given the IUPAC consensus sequence in part 1, what steps would you take to end up with the PWM below? Again, take the IUPAC consensus sequence one letter at a time.

Convert your answer to part 5 into a function. Can you come up with some additional IUPAC consensus sequences to test your code? Try to vary the length of the IUPAC consensus sequences, and make them in uppercase, lowercase, or a mix of both cases. Modify your function so that it can handle all of these cases.
Go back and read the exercise carefully. To answer this exercise properly, there is one additional condition that your function should satisfy: it should ignore any non-IUPAC character. This is not a very specific requirement, so you can interpret this a few different ways. As an example,
Jis not an IUPAC DNA code. So if you have a test sequenceAAJTT, then you can code any of the following (depending on your interpretation) and that would be alright:give an error message, e.g. “this is not an IUPAC DNA code!”
literally just ignore that nucleotide, so give the PWM as if the sequence is
AATTkeep that nucleotide-position, but it would have 0% chance of being either A, C, G, or T, so give the PWM as if the sequence is
AA-0-TTany other way that you interpret this would be fine, just code it so that Python does not throw an error.
Exercise 8.6: Analysis of a regulatory network
For this question use the regulatory network represented in the following dictionary:
hsc_network = {
"Scl" : ["Scl","Gata1","Gata2","Hhex","Zfpm1","Fli1","Erg","Smad6","Runx1","Eto2"],
"Pu.1" : ["Pu.1","Runx1","Gata1"],
"Runx1" : ["Runx1","Pu.1","Erg"],
"Smad6" : ["Runx1"],
"Eto2" : ["Erg"],
"Gata1" : ["Gata1","Scl","Pu.1","Gata2"],
"Fli1" : ["Fli1","Runx1","Pu.1","Scl","Gata2","Hhex","Erg","Smad6"],
"Erg" : ["Smad6","Runx1","Erg","Hhex","Gata2","Fli1"],
"Zfpm1" : ["Gata2"],
"Gata2" : ["Zfpm1","Runx1","Smad6","Eto2","Scl","Gata2","Hhex","Fli1","Erg"],
"Hhex" : ["Gata2"]
}
Each key is a transcription factor and each value is a list of target genes.
Write a function common_targets that accepts three arguments: a TF-Target dictionary, and two TF
names. The function should return a list with all the targets that are shared between the two TFs. The
target list should be alphabetically ordered. When there are no common targets the function should return
an empty list.
With the network above, the following code:
print(common_targets(hsc_network, "Scl", "Gata2"))
print(common_targets(hsc_network, "Smad6", "Eto2"))
print(common_targets(hsc_network, "Zfpm1", "Hhex"))
Should result in:
['Erg', 'Eto2', 'Fli1', 'Gata2', 'Hhex', 'Runx1', 'Scl', 'Smad6', 'Zfpm1']
[]
['Gata2']
If you are struggling with this exercise, click on the triangle to view tips.
To tackle this exercise, let’s start with something a bit more intuitive. Let’s take this dictionary:
room_furniture = {
'bedroom': ['bed', 'lamp'],
'office': ['lamp', 'desk', 'chair'],
'dining_room': ['lamp', 'table', 'chair']
}
As you can see, in this dictionary, each key is a room, and each value is a list of the furniture in the room.
Can you pull out the list of furnitures in the
bedroom? Put it in a list calledfurniture_room_1and print this to the screen.Pull out the list of furnitures on the
officeas well, and put it in a list calledfurniture_room_2and print this to the screen also.Now we are going to ask: what furnitures are common between
furniture_room_1andfurniture_room_2? Let us start by considering the items infurniture_room_1, one by one. Do not code anything yet - just think along. What is the first item infurniture_room_1? Is it present infurniture_room_2? What is the second item infurniture_room_1? Is it present infurniture_room_2?Now try to formulate part 3 in code. What would you use to go over each item in
furniture_room_1, one by one? What would you use to evaluate whether something is infurniture_room_2or not? For now, just print the common furnitures on the screen. (You should end up with justlampon the screen. )Next, instead of printing the common furnitures on the screen, modify your code from part 4 and put the common furnitures in a
list. To do this, first initialize an emptylistcalledcommon_furnitures. Then, every time you identify a common furniture betweenfurniture_room_1andfurniture_room_2, add it to the existinglist.Now let’s turn this code into a function. Name the function
find_common_furnitures. This function should take three arguments:room_furniture_dict,room_1, androom_2. So if you run:find_common_furnitures(room_furniture, 'bedroom', 'office')
It should return a list, and the list should contain a single item ‘lamp’.
Try to run this:
find_common_furnitures(room_furniture, 'dining_room', 'office')
What is the output? Why are the items in the output ordered that way? Modify your code so that the output is in alphabetical order. (Hint: review Chapter 2 if you’ve forgotten how to do this. Alternatively you can just Google it and check out some online guides.)
Now go back to the exercise and read it carefully. Here you are given a dictionary (
hsc_networkinstead ofroom_furniture), where the keys are TFs (instead of rooms), and values are target genes (instead of furnitures). You are asked to find common target genes (instead of common furnitures), when you are given 2 TFs (instead of 2 rooms) and a dictionary. Sounds like something that you can do!
Note that your function “find_common_furnitures” will actually just directly work for this exercise. (Python doesn’t understand rooms or TFs or furnitures or target genes - it is just looping through lists and matching strings.) However, it is important that your functions and variables have names that make sense for what you’re doing, so make sure you modify your code to reflect this.
Exercise 8.7: FASTA statistics
Recall what you have learned about FASTA files in Chapter 7.
Write a function called nuc_fasta.
This function should accept the name of a FASTA file as argument, read the FASTA sequences in that file, and print the nucleotide content of all the sequences.
The function should print a header line, followed by the FASTA id (the sequence name) of every sequence, followed by the nucleotide content of that sequence. The output should be tab-separated. The nucleotide content should be specified as a fraction of the sequence length, with two digits in the order A, C, G and T. When you run your function on the input file /content/gdrive/MyDrive/CFB_files/sequences.fa, the output should exactly mach the following:
name A C G T
chr14:89352059-89352259 0.31 0.28 0.20 0.22
chr5:74264624-74264824 0.34 0.20 0.20 0.26
chr2:132500203-132500403 0.23 0.12 0.21 0.43
chr6:30630663-30630863 0.28 0.23 0.27 0.22
chr15_KI270905v1_alt:1999423-1999623 0.35 0.22 0.15 0.28
Tip 1: If you code is displaying 0.2 instead of 0.20, try out the following code and adapt it for your function:
x = [0.2, 0.2034]
for i in x:
print(i, '\t', format(i, '.2f'))
Tip 2: In Exercise 7.1, you have written a function called read_fasta that reads a FASTA file and returns a dictionary. You can modify (parts of) your code in read_fasta to answer Exercise 8.7.
Exercise 8.8: motif scanning
Write a function that analyzes a FASTA file containing ChIP-seq data, by comparing the sequences in the FASTA file to an IUPAC consensus sequence (i.e., a motif). As an optional argument, a user should be able to specify a number of mismatches. As output, the function should return, for every match: (i) the ID of the sequence, and (ii) the position of the match within the sequence. For (ii), this should be the index of the starting position of the match within the sequence in the FASTA file, not the position of the sequence on the chromosome.
Before you begin, you may want to refresh your knowledge of ChIP-seq and motifs by reviewing the lecture materials.
Test data
You can use the following files (adapted from Chen et al. 2008), located in /content/gdrive/MyDrive/CFB_files, to test your code.
consensus.txtc-Myc.faSTAT3.fa
Please note: when you are just starting to answer this question, it may be beneficial to simply open the file consensus.txt and copy-pasting a sequence from this file directly into your code. This may help you write (and test) your code. However, in your final answer, your code should be reading the consensus sequences from this file computationally.